Butler Blue Downloads
Download your favorite Butler Blue activities.
Coloring Sheets
- Color Blue!
- Color in the Basketball
- Design your own jersey
- Trace and Color the Court
- Color the Butler Bulldog
Holidays
Puzzle Pages
Holidays
Homecoming Color Contest
Calling all artists! Our favorite artist and illustrator of Good Boy, Blue, Jingo de la Rosa, has put together this simple step-by-step guide to learn how to draw Blue!
Click picture to download the different wallpapers.
Butler University |
     |
Butler Blue IV |
    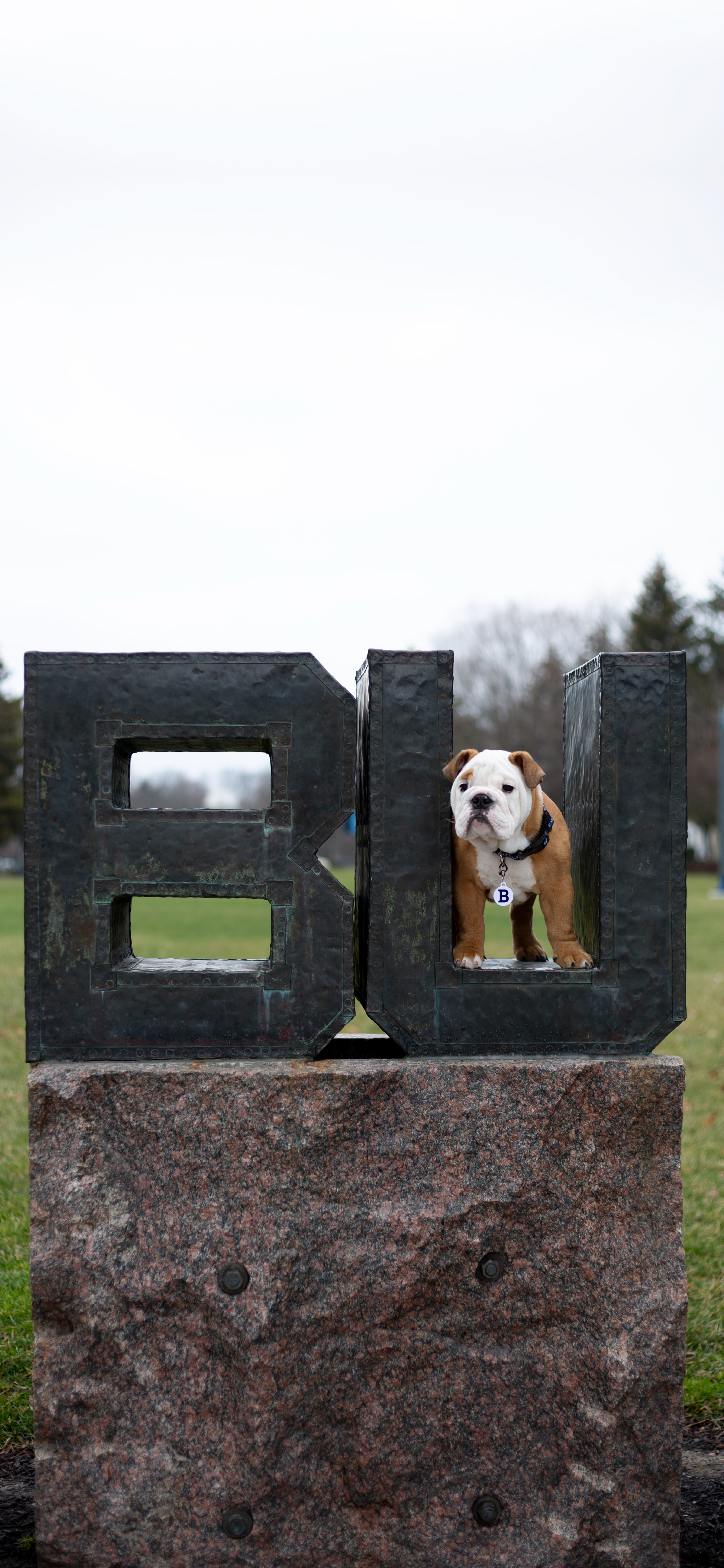     |
Butler University |
      |
Butler Blue IV |
            |
Hinkle Fieldhouse |
    |
Click picture to download the different wallpapers.
Butler Blue IV



There is no better way to swoon the one you love more than to use my face. Download the Butler Blue IV Valentine’s Day cards here!
Add some Butler flair to your porch this Halloween season! You can download the official Butler Blue Jack-O-Lantern Stencils here!
Sit back, grab a cup of cocoa and enjoy as I nap and chew a bone next to a roaring yule log anxiously awaiting Santa’s arrival. Wait did you hear that too …?
Watch here!